Cell death mechanisms can be categorized into several types, including apoptosis, necrosis, autophagic cell death, and regulated forms like ferroptosis and disulfidptosis. Each type of cell death has distinct biological functions and characteristics.
Disulfidptosis is a form of regulated cell death mediated by disulfide stress, which arises from excessive cystine uptake and impaired NADPH generation.
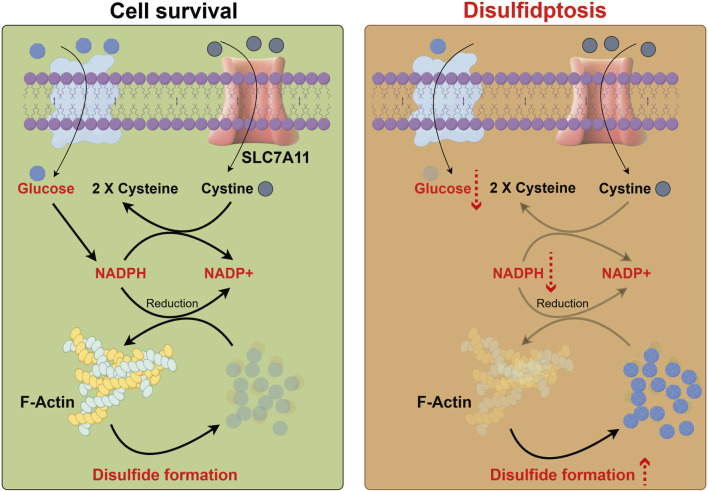
Fig 1. Process diagram of disulfidptosis (Front Cell Dev Biol. 2025 Jun 4;13:1559423.)
The occurrence of disulfidptosis may require the convergence of three key features:
High expression of SLC7A11: This protein mediates the exchange of extracellular cystine for intracellular glutamate, leading to excessive cystine uptake and abnormal intracellular accumulation of cysteine, thereby inducing disulfide stress in metabolism.
Glucose starvation: This condition inhibits the pentose phosphate pathway (PPP), impairing NADPH production.
Formation of aberrant disulfide bonds between actin cytoskeleton proteins.
When these conditions are met, excessive disulfide accumulation promotes disulfide cross-linking between actin cytoskeletal proteins. This triggers actin contraction and detachment from the plasma membrane, ultimately causing cell shrinkage and death. This process is not inhibited by traditional apoptosis or necrosis inhibitors, representing an independent cell death pathway.
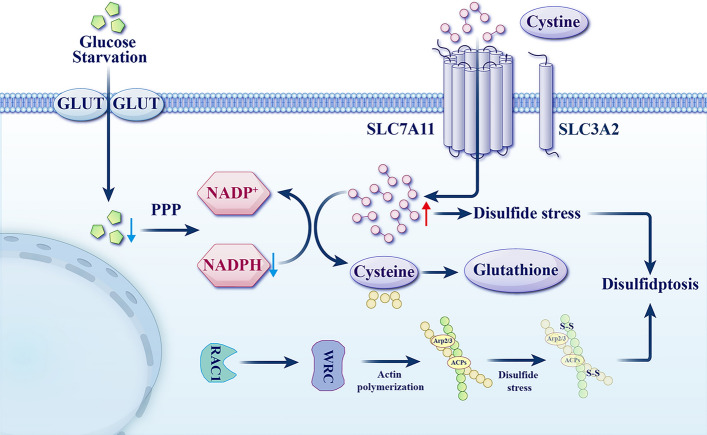
Fig 2. Schematic diagram of disulfidptosis mechanism in SLC7A11 overexpressed cells (Cell Mol Biol Lett. 2025 Jun 2;30:66.)
Notably, if low SLC7A11 expression coincides with glucose starvation or uptake blockade, intracellular glucose levels decrease, inhibiting hexokinase-mediated production of glucose-6-phosphate. This subsequently impedes both NADPH generation via the PPP and pyruvate production via glycolysis. These reactions further suppress pyruvate metabolism in the tricarboxylic acid (TCA) cycle and mitochondrial oxidative phosphorylation, ultimately inducing oxidative stress and ATP depletion. This process involves upregulation of Bax/Bak expression, promoting cytochrome c release from mitochondria, activation of caspase-3, and PARP-mediated membrane blebbing, leading to apoptosis.
Conversely, if high SLC7A11 expression occurs alongside glucose starvation or uptake blockade, it results in massive cystine uptake and its reduction to cysteine, concomitant with NADPH depletion and intracellular disulfide accumulation, ultimately inducing disulfide stress. This stress state, through activation of the Rac–WRC–Arp2/3 signaling pathway, promotes the formation of aberrant disulfide bonds in actin cytoskeletal proteins, thereby triggering disulfidptosis.
These disulfide bonds form cross-links between actin, MYH9, and at sites like c988-c1379, detectable by characteristic band shifts in electrophoresis. Morphologically, this is characterized by the collapse of the F-actin network and cell contraction, culminating in the detachment of F-actin from the cell membrane. Further analysis reveals that regulators such as the RAC1–WAVE complex and NCKAP1 participate in actin network remodeling; knockout of NCKAP1 significantly delays the progression of disulfidptosis, indicating that dynamic regulation of the cytoskeleton is a key node in this process.
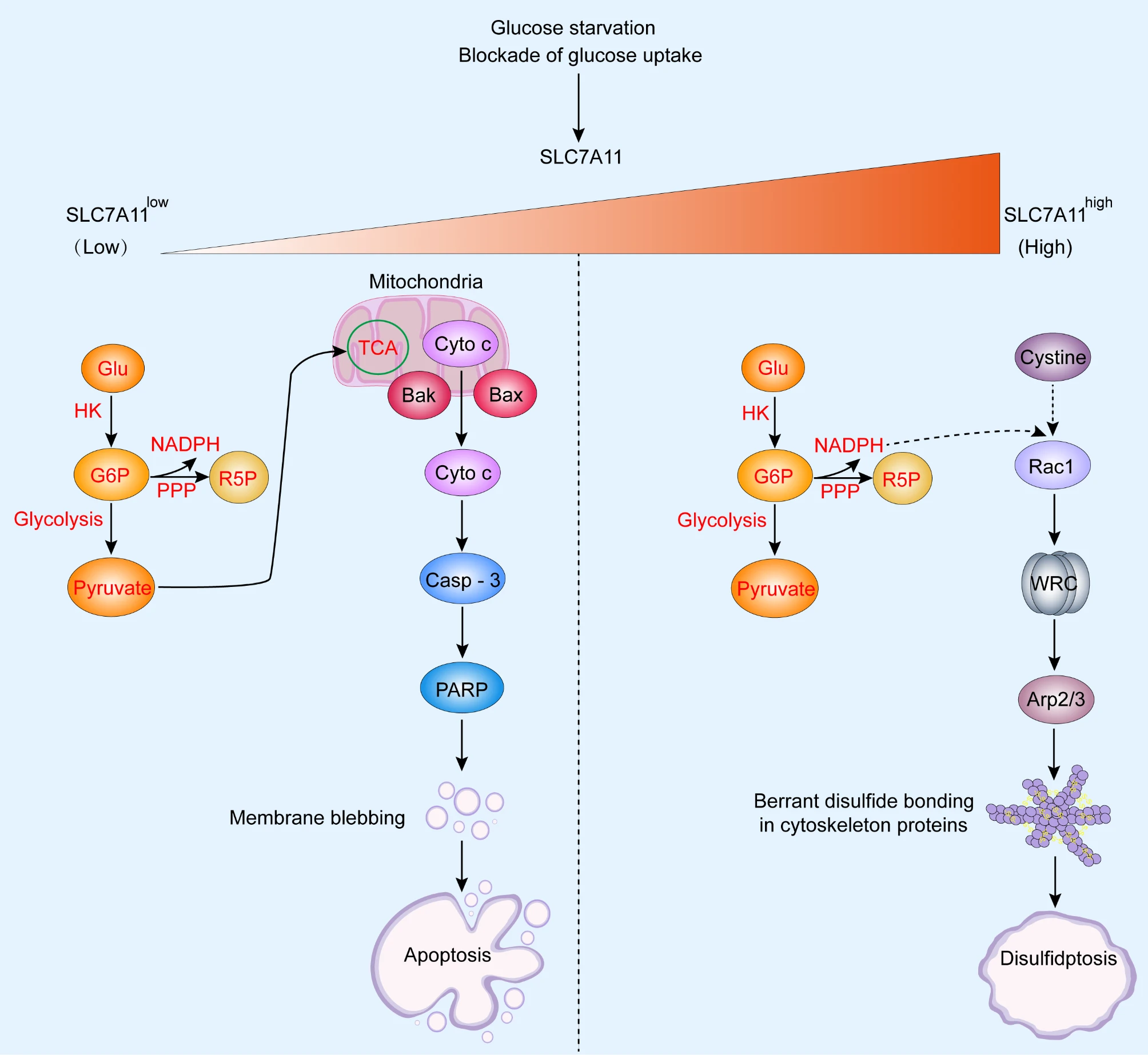
Fig 3. Schematic overview of the main difference between apoptosis and disulfidptosis (J Exp Clin Cancer Res. 2023 May 31;42(1):137.)
Recent studies have found that under glucose starvation, SLC7A11-mediated cystine uptake, while inhibiting ferroptosis, can also induce disulfidptosis. Although both are related to System Xc− function, their mechanisms differ significantly: Disulfidptosis is primarily characterized by an increased NADP+/NADPH ratio and disruption of the disulfide/NADPH balance, whereas ferroptosis mainly involves the accumulation of lipid peroxides.
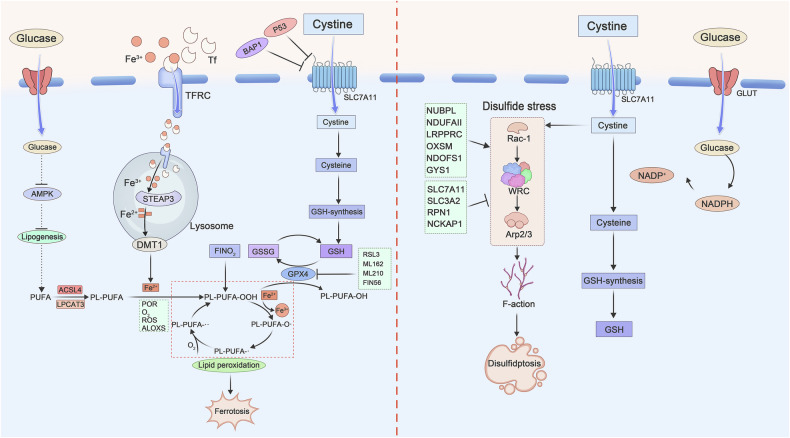
Fig 4. Molecular mechanisms of ferroptosis and disulfidptosis (Cell Death Discov. 2025 Apr 28;11:205.)
Disulfidptosis is closely associated with the pathogenesis and progression of various diseases, particularly cancer. Studies show that tumor cells with high SLC7A11 expression are highly susceptible to disulfidptosis under glucose deprivation or GLUT inhibitor treatment, providing a potential synthetic lethality strategy for metabolism-targeted therapy. Genes associated with disulfidptosis have been confirmed to have prognostic value in solid tumors such as gastric cancer, hepatocellular carcinoma, and oral squamous cell carcinoma. Related long non-coding RNAs can also regulate their expression and influence patient survival.
Although certain mechanisms of disulfidptosis are not yet fully elucidated, its potential in cancer therapy is evident. Inducing disulfidptosis in cancer cells offers two significant advantages: Firstly, the process relies on specific metabolic characteristics, potentially allowing selective targeting of cancer cells without harming normal cells, thereby reducing side effects. Secondly, tumors resistant to conventional therapies might be more prone to disulfidptosis. For instance, some cancer cells that highly express SLC7A11 to resist apoptosis and ferroptosis develop chemoresistance. In such SLC7A11-high tumors, glucose deprivation can rapidly induce disulfidptosis, while normal cells exhibit stronger tolerance. Currently, inducing disulfidptosis has emerged as a promising new strategy in cancer treatment.
Furthermore, disulfidptosis has been reported to contribute to the progression of non-alcoholic fatty liver disease (NAFLD), where disulfide stress-induced destruction of the hepatocyte cytoskeleton accelerates fibrosis and disease progression. Preliminary research also suggests that disulfidptosis may play a regulatory role in neurodegenerative diseases such as Alzheimer's disease and Parkinson's disease, although the specific mechanisms require further exploration.
In summary, as an emerging form of cell death, disulfidptosis, with its unique disulfide stress mechanism, shows broad application prospects in metabolic cancer therapy and metabolic disease research. Future work should focus on precisely deciphering its molecular network, developing drugs targeting the SLC7A11–NADPH axis, and clinically validating its feasibility as a biomarker and therapeutic target.
Below is an overview of several common cell death modalities:
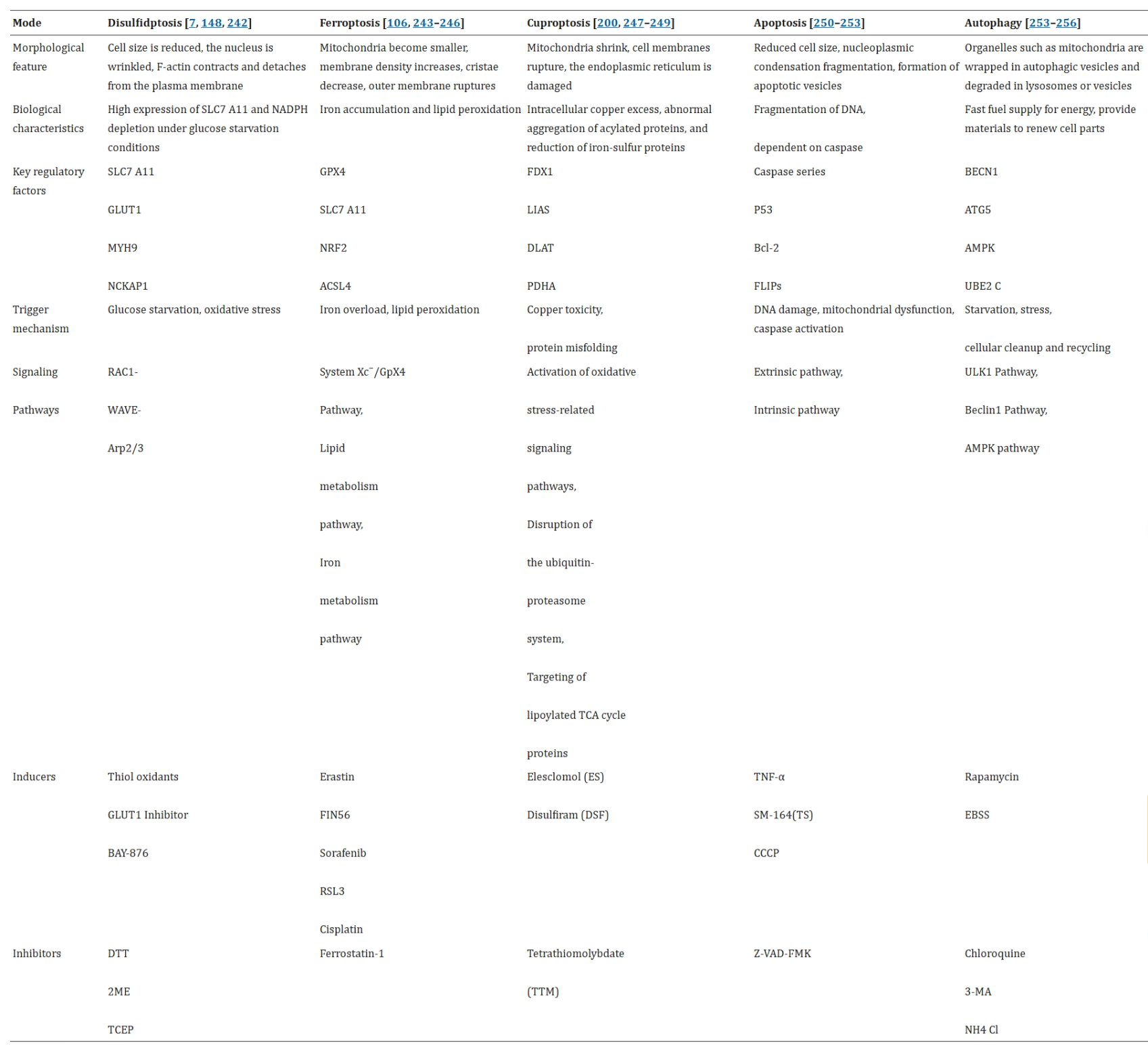
Fig 5. Different Cell Death Modes (Cell Mol Biol Lett. 2025 Jun 2;30:66.)
References
1. Liu Y, Li S, Wu Y, et al. Molecular signatures of disulfidptosis: interplay with programmed cell death pathways and therapeutic implications in oncology. Cellular & Molecular Biology Letters. 2025;30(1).
2. Zhu WW, Liu Y, Yu Z, Wang HQ. SLC7A11-mediated cell death mechanism in cancer: a comparative study of disulfidptosis and ferroptosis. Frontiers in Cell and Developmental Biology. 2025;13.
3. Zheng T, Liu Q, Xing F, Zeng C, Wang W. Disulfidptosis: a new form of programmed cell death. Journal of Experimental & Clinical Cancer Research. 2023;42(1).
4. Wan S, Liang C, Wu C, et al. Disulfidptosis in tumor progression. Cell Death Discovery. 2025;11(1).
AntibodySystem provides Disulfidptosis-related products, delivering more tools and solutions for research.
|
Catalog |
Product Name |
|
RHK09202 |
Anti-SLC7A11/xCT Antibody (R2L62) |
|
RHK09201 |
Anti-SLC7A11/xCT Antibody (R2L63) |
|
PHE13201 |
Anti-MYH9 Polyclonal Antibody |
|
RHE13202 |
Anti-MYH9 Antibody (R1V12) |
|
RHB85001 |
Anti-LDHA Antibody (R1B36) |
|
RHB85003 |
Anti-LDHA Antibody (R1H05) |
|
RHB85002 |
Anti-LDHA Antibody (R1H06) |
|
RHB85004 |
Anti-LDHA Antibody (R1H07) |
|
PHC85901 |
Anti-Human SLC2A1 Polyclonal Antibody |
|
RHC86102 |
Anti-SLC2A3/GLUT3 Antibody (R1N27) |



